METRAILER, JIRANONTASOK, LUONG, HAMERLINCK, NORRIS, BLACKBURN – Spatial and phylogenetic patterns reveal hidden infection sources of Bacillus anthracis in an anthrax outbreak in Son La province, Vietnam
Morgan C. Metrailer, Thi Thu Ha Hoang, Treenate Jiranantasak, Tan Luong, Luong Minh Hoa, Do Bich Ngoc, Quang Thai Pham, Van Khang Pham, Tran Thi Mai Hung, Vu Thi Lan Huong, Thanh Long Pham, Jos ́e Miguel Ponciano, Gabriela Hamerlinck, Duc Anh Dang, Michael H. Norris, Jason K. Blackburn
Article first published online: 7 September 2023
DOI: https://doi.org/10.1016/j.meegid.2023.105496
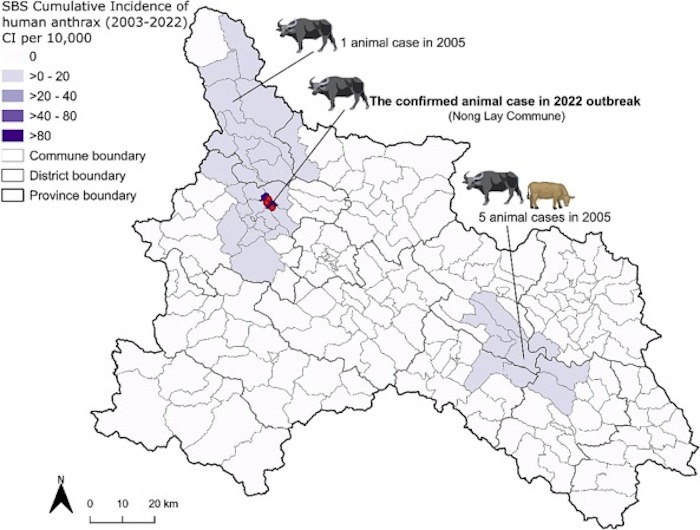
ABSTRACT: Bacillus anthracis, the bacterial cause of anthrax, is a zoonosis affecting livestock and wildlife often spilling over into humans. In Vietnam, anthrax has been nationally reportable since 2015 with cases occurring annually, mostly in the northern provinces. In April 2022, an outbreak was reported in Son La province following the butchering of a water buffalo, Bubalus bubalis. A total of 137 humans from three villages were likely exposed to contaminated meat from the animal. Early epidemiological investigations suggested a single animal was involved in all exposures. Five B. anthracis isolates were recovered from human clinical cases along with one from the buffalo hide, another from associated maggots, and one from soil at the carcass site. The isolates were whole genome sequenced, allowing global, regional, and local molecular epidemiological analyses of the outbreak strains. All recovered B. anthracis belong to the A.Br.001/002 lineage based on canonical single nucleotide polymorphism analysis (canSNP). Although not previously identified in Vietnam, this lineage has been identified in the nearby countries of China, India, Indonesia, Thailand, as well as Australia. A twenty-five marker multi-locus variable number tandem repeat analysis (MLVA-25) was used to investigate the relationship between human, soil, and buffalo strains. Locally, four MLVA-25 genotypes were identified from the eight isolates. This level of genetic diversity is unusual for the limited geography and timing of cases and differs from past literature using MLVA-25. The coupled spatial and phylogenetic data suggest this outbreak originated from multiple, likely undetected, animal sources. These findings were further supported by local news reports that identified at least two additional buffalo deaths beyond the initial animal sampled in response to the human cases. Future outbreak response should include intensive surveillance for additional animal cases and additional molecular epidemiological traceback to identify pathogen sources.
Read the full publication in Infection, Genetics and Evolution.